|
Cyclooctane.world File Reference Detailed Description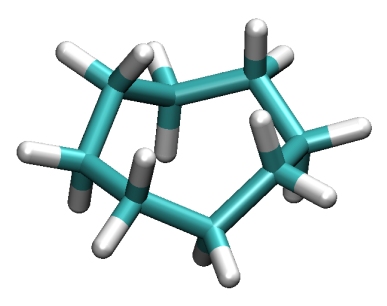
IntroductionCyclooctane is a cyclic alkane used as an intermediate in production of plastics, fibers, adhesives, and coatings. It is also known as octomethylene. The goal here is to compute an exhaustive map of all conformations that are compatible with the rigid-geometry hypothesis (fixed bond lengths and angles, variable torsion angles). To the best of our knowledge, a complete map of such conformations was not known so far. Other previous work on this molecule could only obtain low-energy conformers via sampling and/or local search techniques [Kolossvary and Guida 1993, Manocha and Zhu 1994, Rocha et al. 1998]. The map we provide is exhaustive (it contains all points of the conformational space) and may thus be used to determine its topology, or derive the whole (guaranteed) network of minimum-energy conformers and saddle point transitions between them. The formulation provided here is not based on distances (as we did in Porta et al. 2007 but on rotation matrices and vectors (as in Porta et al. 2008). In any case, this problem is very hard and we use a cluster of computers to isolate the whole configuration space. The difference is that with the distance formulation days of compuation are necessary, even when using the cluster, and with the vector/matrix formulation few minutes are enough. ProcessThis example is treated following this steps (from the main CuikSuite folder):
Please, see the experiments with molecules for additional information about how to deal with the cyclooctane. StatisticsCharacteristics of the problems:
Here you have the statistics about the execution (on a grid with 160 cores).
ResultsThe conformational space of the cyclooctane, shown in transparency so that its complexity can be appreciated. 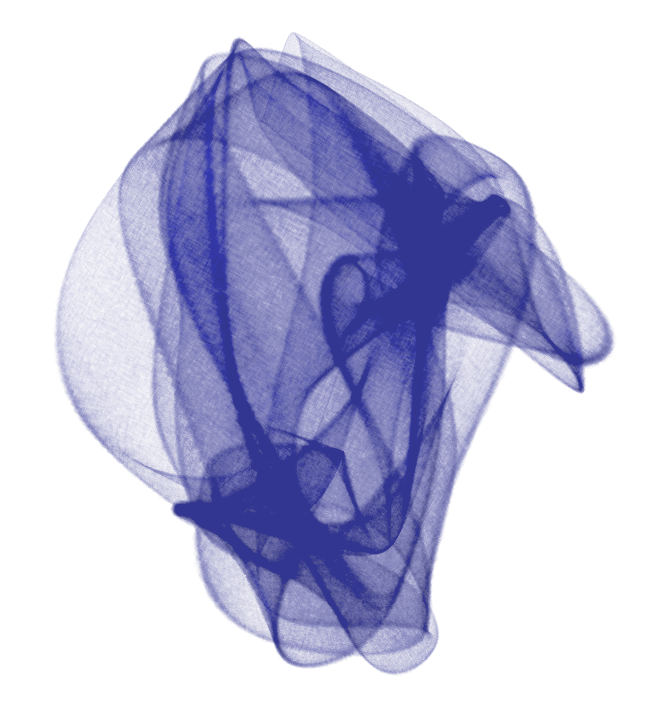
The solution of cuik set includes about 280000 boxes, so the process and visualization would be slow. This is the atlas obtained with bin/cuikatlas and colored using the potential energy of each conformation. 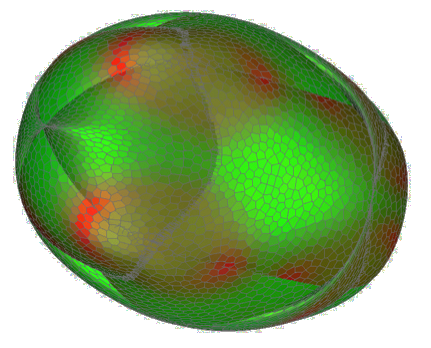
Please, see our experiments with bio molecules for more information about this how to obtain this output. Note that the atlas and path planning process can also be directly applied to the world file, but then collisions are taken into account. Note that the result is a two-dimensional manifold and therefore it is not worth to use tools such as cuiksort or cuikanimate on the solution file (these tools are basically aimed for zero or one-dimensional solution sest). Morever, you can also get particular configurations (using cuiknewton) and determine a path between them using cuikatlasGBF or cuikatlasrrt. These applications can work on different representations for the problem but they are typically more efficient using a DOF-based representation (set REPRESENTATION parameter to DOF). In this case the samples to connect are read from the dof file. Note that conversion between sample files (containing configuraions in MATRIX representation) and dof files are possible using cuiklinks2joints. It is important to remark that the obtained path does not take into account energetic aspects but only kinematic ones. Please note that different samples/dof are provided for the cyclooctane corresponding to different set ups for this problem used in our papers. The difference between the experiments is the value of rH (the radii for the hydrogen atoms causing collisions) set in the Cyclooctane.world file. References
Definition in file Cyclooctane.world. |
Follow us!